
This sub-option allows estimation of gene frequencies when a null allele is present. Different methods are available: maximum likelihood, maximum likelihood with genotyping failure, and Brookfield's (1996) estimator, which differences are explained in Appendix 1.
Genepop takes the allele with the highest number for a given locus across all populations as the null allele.20 For example, if you have 4 alleles plus a null allele, a null homozygote individual should be indicated as e.g. 0505 or 9999 in the input file.
The default estimation method is maximum likelihood, using the EM algorithm of Dempster et al. (1977). Apparent null genotypes may also be due to nonspecific genotyping failures. Joint maximum likelihood estimation of such failure rate ('b') and of allele frequencies is available by selecting the appropriate option on the input form. The estimator of Brookfield is also available for selection on the input form. Confidence intervals for null allele frequencies are computed for each locus in each population. Their coverage probability cannot be modified on the web version, however it is possible to alter these confidence intervals using the PC version of Genepop available from http://kimura.univ-montp2.fr/~rousset/Genepop.htm.
The output file for this option may contain:
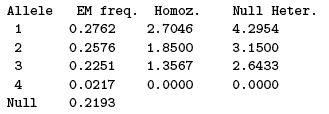
Note that there may be insuffcient information to compute estimates and/or confidence intervals: not enough alleles in the sample, for example. These are indicated by the message No information. Sometimes the point estimate can formally be computed but the computed CI is not meaningful. This happens for example in case of heterozygote excess, and generates a (No info for CI) warning (if all pseudo-samples generated by some resampling technique show an heterozygote excess, all pseudo-estimates of null allele frequency will be zero and there is no information to construct a non-null CI from this distribution).
This sub-option "diploidizes" an haploid data set. For example, the line
popul 1, 01
02 10 00
of an haploid dataset with 4 loci, will become
popul 1, 0101 0202 1010 0000
Only haploid data are thus modified in a mixed haploid/diploid datafile. The new data set is returned via the web browser (or email) which you can save as a text file.
Note that there may no longer be any need for this option for further analyses with Genepop (except perhaps as a preliminary to le conversions, option 7), since Genepop 4.0 now performs analyses on haploid data without such prior 'diploidization'.
Results for all sub-options are returned via your web browser which you can then save to you local machine. You may also choose to have them emailed to you.
Last Modified on December 1, 2020 by Eleanor Morgan